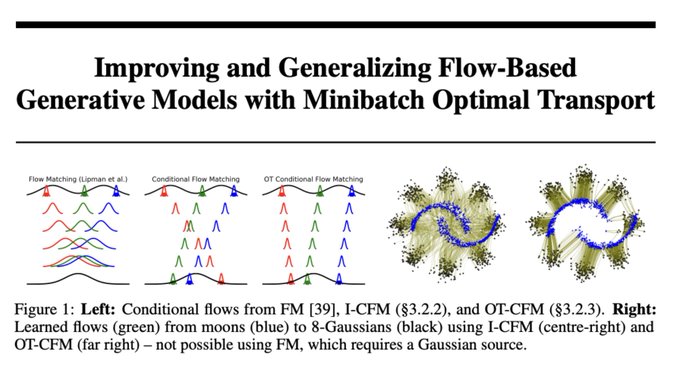
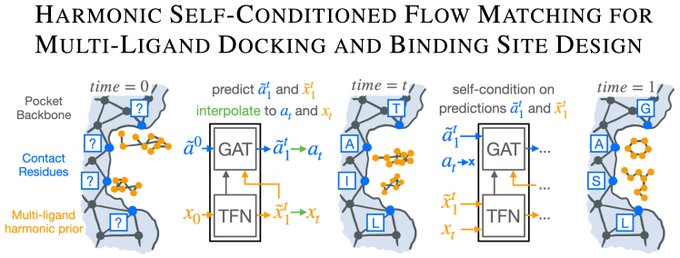
Alex Tong
@AlexanderTong7
2,235
Followers
521
Following
6
Media
133
Statuses
Postdoc at Mila studying cell dynamics with Yoshua Bengio. I work on generative modeling and apply this to cells and proteins.
Don't wanna be here?
Send us removal request.